ȫ���������������ִ����������������ڷ�֧��science��2014-12-12��
ժҪ
Ϊ�˸��õ�ȷ���ִ��������ʷ����������Ϊ�����������ģ���ݶ�������ϵͳ������ѧ�������Դ�������������Ŀ��48�����ֽ����˻������ģ��ϵͳ���������������ҵ���һ�ø߷ֱ��ʵ�������֤ʵ����ǰ������Ľ��û����ܹ�ϵ������ȷ����������Ŀ�ĵ�һ����֧�����dz�֮Ϊȸ����࣬���Ǵ����˲�ͬ�ġ��ۺ��ݻ���½�غ�ˮ�����ֵĶ���Ѫͳ����ȸ�ද���У������ƶϺ���½�ݵĹ�ͬ������һ�ֶ�����ʳ�ߣ�����֤ʵ�˷���ѧϰ����Ķ�����չ���ڸ��ද���У������ƶϸ��Ӻͻ����������ڽ��ý���֦����ʹ�������������飬������Ŀ�������һЩ��֧����ȥ�ֱ棬����õĽ����Ǵ����ĵ����ʱ������л�ۺ߶Ȳ���ȫ����ϵ���࣬���Ƿ����ڴ�Լ6600����ǰ���Ѽ�-�Ž��ʹ�����¼����һ�ο��ٷ����С�
���ֵĶ����Բ������ǽ���ʽ�ģ�Ҳ�ᷢ���ڿ��ٵķ����У��ر��Ǿ������ش�Ļ����仯֮������ѧ�ͷ�������ѧ֤�ݱ�����Щ��ը�����䷢����������Ŀ�ϣ����� ���ݡ����ġ����ӵ������ģ�����̥�̵IJ��鶯�ռ�ִ�����Ͳ��鶯�����ֵ�95%���ھ���Լ6600����ǰ��Ma��megaannus���������꣬��10��6�η��꣬����ѧ��������ѧ�������õ�ʱ�䵥λ����ʱҲд��Myr�����İ��Ѽ����Ž���(K-Pg)�Ĵ�����¼���Ȼ��������ԭ�Ӻ˺�������DNA���о��������ھ��80-125������ǰ��ʼ�˽���ʽ�Ķ����ԡ���ͬ�����ݼ��ͷ�����������������ͬ�ĵ����ֽṹ�������Է��ֻ�����������顣���������ʱ���ϵͳ������ϵ�ԱȽϻ�����ѧ�dz���Ҫ����Ϊ�Ƚϻ�����ѧ���Խ�ʾ����������ͼ�����
����������о�����5[~5000����ԣ�bp��]��19[31000bp]����Ƭ�Σ��ָ���һЩ����̬ѧ���ݺ�DNA-DNA�ӽ��ƶϵĹ�ϵ���������µĹ�ϵ����������������ϵ��ì�ܡ�������֮ǰ�ķ��Ӻ͵�����̬ѧ�о�һ�£����ǰ��ִ����ࣨ�����ࣩ���ֳ������࣬�ֱ��ǹ���¸٣�?��Ŀ�Ͳ��ܷɵ�ƽ���ࣩ����ȸС�١�����Ŀ��½�ݣ�������Ŀ��ˮ�ݣ���������С�٣������������ִ����ࣩ��������С�٣���������˼����µĴ���Ⱥ������һ����������졢���ɡ�DZ���ˮ�����Ⱥ����һ֦��������ľ�����ݡ����ĺ����ݵ�½�����Ⱥ��������������ЩŬ����������С��������ķ�֧֮��Ĺ�ϵ��һЩ���ھ�����ս�Եķ���Ⱥ��λ�ã�����������^�����ص���������������С�ٵĵ�һ�η�֧�ļ���������ֳ�����ͬ����ķ�֧����ΪMetaves��Coronaves����û�н����
��Ȼ����Ķ�����о���һЩ�����Ѿ�����������л���ת�����Ӳ������ݼ�֤ʵ�ˣ����������������֦��û�еõ�֧�֡����⣬������������ȫ����������ָ��˲�ͬ�Ĺ�ϵ��������֧�ָ���½��ĵ�ϵ�ԡ��о��е�һЩ��ͬ���������ڻ������IJ�һ�£�������������Щ����IJ���ȫ��ϵ���࣬�����������ƫ���������֮��IJ��죬���������ݲ�����ɵġ���ˣ�����ȷ���ض���������������ѧ��ѧϰ���Ӷ���Ϊ�������Ƕ�ˮ��������½����������Ӧ���Ƿ�ӳ�˵������߶����������Դ���Լ��ں�����̬�����·�������Щ�¼���
һ���ձ�ļٶ��ǣ�����ÿ�����ֵĻ������д����������ݻ���¼����ǿ��ͳ��������ȫ���������ݽ�����ϵͳ�����ؽ�������ͨ�����ռ�����װ��48������������ϵͳ������������֤��һ���裬��Щ������������ִ�����й��ϵ�������Ŀ������������Ŀ�����а���һЩ���������ж�������ࡣ
����ѡ����������ķ�չ���Լ����������ݼ�����֤��
���Ǹ��ݲ�ͬ�ķ��ʽѡ�����ܴ�������������Ŀ�����֡����ǰ�������Щ�Ѿ����ѷ��������֧����Ⱥ������˵������������ҹӥ���������ȡ��кס�����Ҳ��������Щ�������Ƚڵ��½������֣����ǵ�˳���Ƿ���DZ�ڵij���֧������˵����Ŀ��ȸ��Ŀ������Ҳ������ѧ�������֣������ࡢ�������ģ������DZ�������������ģ�ͣ��Լ��������������Ƶ���ѧ������Ե��ȡ���������������ࡢ���ࡢ���������Ǿ飩�����������֮��IJ�ͬ���Ӷ������Ǹ��Զ����Ļ����еó���ͬ�Ľ��ۡ����õ��Ľ�����ݼ�����45����������飬����һ������Ϊ����Ŀ������֮ǰ����������ʱ��48���������ַ��������ж���������ꡢ�̺���������桿
������������ǰ��С��ģϵͳ�������о���û�������ļ�����ս�����в�ͬע�͵Ļ�����ʹֱϵͬԴ��ļ�����ø��ӣ��������ݾ���Ĵ�Сʹ����������ʹ�����������ϵͳ�������ߡ�Ϊ���跨�����Щ��ս�����ǻ��ڼ��Ͱ���ȸ�Ļ���Ⱥ�Զ�������������������˵����ʱ�������ͳһע�͡����Ƿ���SATe�����Աȳ���������㷨�Դ��ģ�����ݲ����˸��ɿ��ĶԱȣ��������ǿ����˶��˲��㷨������δ����ʹ����������С����ǿ�����ExaML�����������Ȼ����RAxML��һ�ּ���Ч�ʸ��ߵİ汾�����ڸ��ݻ�����߶ȼ������жԱȹ���������������Ҳ������һ��ͳ�Ʒ��䷽�����Ľ��˶����ֺϲ��������Դ������е�ϵͳ�����źŵĻ��������ƶ�����������Щ�����ܼ��ͷ�������9�����ϵij������Ľ��еģ���Ҫʹ�õ�����������400�����ϵļ��㡣
ͨ����ЩŬ��������ȷ���˿�����ĸ�������ֱϵͬԴ�����飬�û�������8251��ͬ�������ʱ������Լռ���������40%���������ӣ�2516����Щ������ں��ӣ��Լ�һ����ص���3769������1000bp�������еij�����Ԫ�أ�UCE������Щ���е�֤�ݺ��������ݼ�����Լ4180�������ԣ�bp=base pair����Լռ����ƽ���������3.5%��
�������ģ������ϵͳ����
��֤�ݺ�������
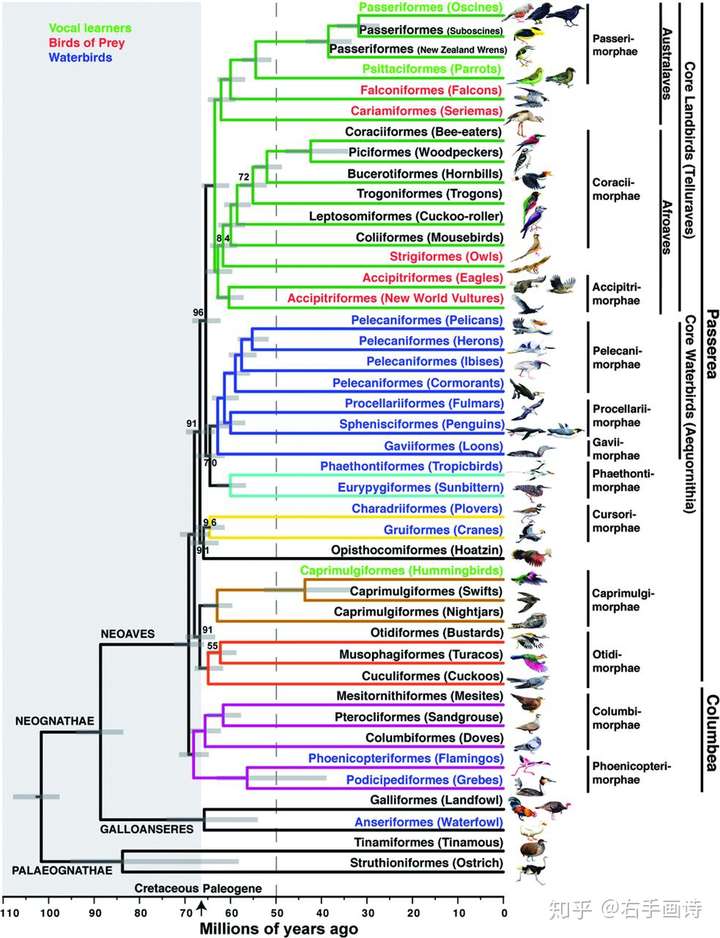
��GTR+GAMMA���н���ģ�ͣ�SM4���£���ExaML�����˰��������ͣ��ں��ӣ�UCE����һ�͵ڶ�������������λ�ã�����λ������������ų������ֵ�ȫ֤�ݺ��������У��õ��˸߷ֱ��ʵ���֤�ݺ���������TENT����������������֧�ֶȣ�BS���İ����£��ָ����ִ������е��������ϵ���Ҫ��Ⱥ����ʼ���¸١���ȸС�١�����С�٣����������ֺϲ��������¸��£����������ʾ���ִ������С���еĵ�һ�����죬���²�����������ȫ֧�ֵģ����ϵ�Ľ��ý���֦�����ǽ�������Ϊȸ�ࣨ�������������Ⱥȸ��Ŀ���������ࣨ�������������Ⱥ����Ŀ��������



 �Ӻ���
�Ӻ���  ������
������
 �۾�
�۾�
 Post By��2021/4/13 13:39:35 [ֻ��������]
Post By��2021/4/13 13:39:35 [ֻ��������]